<em>Gesetzgebung</em>
Die neue europäische Medical Device Regulation (MDR 2017/745, EU-Medizinprodukte-Verordnung) und die In-vitro Diagnostic Regulation (IVDR 2017/746, EU-Verordnung über In-Vitro-Diagnostika) ersetzen die bestehenden Medizinprodukte-Richtlinien.</p> <p><strong> </strong></p> <ul> <ul> <li>die <strong>MDD 93/42/EWG </strong>und die<strong> AIMD 90/385/EWG </strong>werden zur<strong> MDR 2017/745
</strong></li> </ul> </ul> <ul> <ul> <li>die <strong>IVD 98/79/EG </strong>wird zur<strong> IVDR 2017/746
</strong></li> </ul> </ul> <p> </p> <p>Seit dem <strong>25.05.2017</strong> sind die EU-Verordnungen, die MDR und die IVDR, in Kraft getreten. Nach einer Verschiebung auf Grund der COVID-19 Pandemie ist nun neu die MDR ab dem <strong>26.05.2021</strong> anwendbar. Die Anwendbarkeit der IVDR wurde wie geplant auf dem <strong>26.05.2022</strong> belassen.
<em>Ziel der MDR / IVDR</em>
Ziel der neuen EU-Verordnungen ist es, die Patientensicherheit weiter zu verbessern und zu erhöhen. Dementsprechend sehen die wesentlichen Änderungen der MDR folgenermassen aus:</p> <p> </p> <ul> <ul> <li>erhöhte Anforderungen an die Qualifikation vom Personal der Prüfstellen, lückenlose Rückverfolgbarkeit der Medizinprodukte unter Mithilfe einer eindeutigen Kennzeichnung</li> </ul> </ul> <ul> <ul> <li>erhöhte Anforderungen an den Nachweis der klinischen Wirksamkeit von Produkten sowie die Registrierung aller Produkte in der zentralen Europäischen Datenbank für Medizinprodukte (EUDAMED).</li> </ul> </ul> <ul> <ul> <li>Verschärfung der Auflagen zur Marktüberwachung.</li> </ul> </ul> <p> </p> <p><strong>Zeitplan</strong>
Die Übergangsbestimmungen und die Übergangsfristen sind in den beiden Verordnungen sehr komplex formuliert. Zudem gab es durch die Verschiebung der <strong>EUDAMED</strong> um 2 Jahre auf den <strong>26.05.2022</strong> und die oben erwähnte Verschiebung der Anwendbarkeit der MDR immer wieder Änderungen.
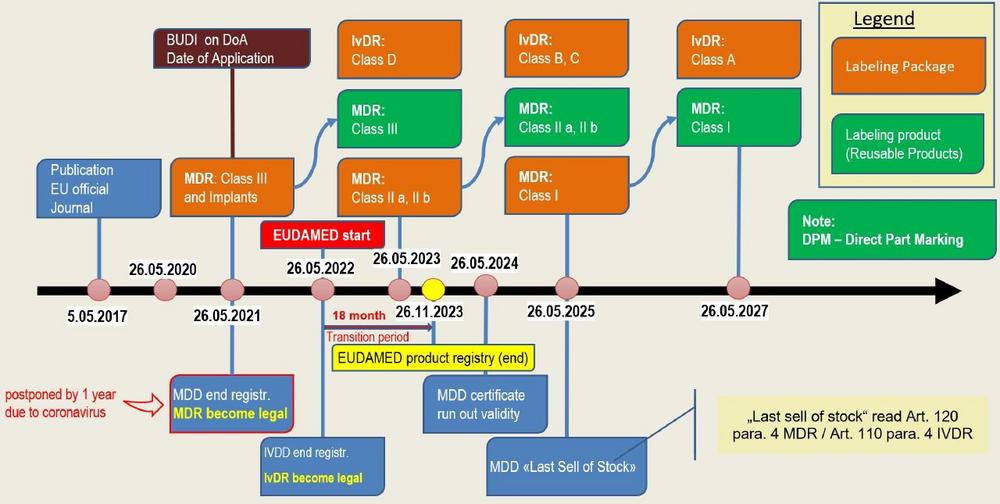
Im folgenden Abschnitt zeigen wir Ihnen mit Hilfe einer Grafik auf, wann welche Gesetzgebung in Kraft tritt bzw. umgesetzt werden muss.
Der Zeitpunkt der Registrierung der Produkte hängt von diversen Faktoren ab. Zum einen hat die Klassifizierung der Produkte einen Einfluss. Zudem wird unterschieden ob es sich beim Produkt um ein direkt markiertes Produkt handelt oder ob die Verpackung etikettiert wird.
Bei wiederverwendbaren Produkten, welche direkt markiert werden, gewährt die Verordnung zwei zusätzliche Jahre. Den genauen Zeitplan sehen Sie im grafisch dargestellten Zeitplan.
<strong>Änderungen</strong>
Die Auswirkungen der MDR sind weitumfassend. Es werden verschiedene Bereiche tangiert was die Medizinprodukte-Industrie vor grosse Herausforderungen stellt. Die wichtigsten Änderungen sind:
<em>Erweiterung des Geltungsbereiches</em>
Die Definitionen im Bereich der Medizinprodukte und aktiven implantierbaren medizinischen Geräte werden erheblich erweitert und umfassen nun beispielsweise auch Produkte ohne medizinische Zweckbestimmung (z. B. farbige Kontaktlinsen, Implantate und Stoffe für ästhetische Zwecke etc.)
<em>Benennung einer „qualifizierten Person“</em>
Medizinproduktehersteller müssen in ihrer Unternehmung mindestens eine Person benennen, die schlussendlich dafür zuständig ist, dass die Anforderungen der MDR erfüllt werden. Die Unternehmung muss nachweisen können, dass diese Person im Besitz der notwendigen Qualifikationen im Hinblick auf die geforderten Aufgaben ist.
<em>Umsetzung des Systems der einmaligen Produktidentifizierung</em>
Die Gesetzgebung fordert ein System der einmaligen Produktidentifizierung welches mit dem System UDI (Unique Device Identification) umgesetzt werden soll. Damit soll die Rückverfolgbarkeit bestimmter Produkte der Lieferkette für Hersteller und Behörden vereinfacht werden. Daraus resultiert ein schnellerer und effizienterer Rückruf von Medizinprodukten, welche ein Sicherheitsrisiko darstellen. In diesem Zusammenhang sollte auch dem Labeling der Produkte Aufmerksamkeit geschenkt werden. Dies muss den Konformitätsanforderungen der MDR entsprechen.
Zudem soll die Europäische Datenbank für Medizinprodukte (EUDAMED) erweitert und benutzerfreundlicher gestaltet werden. Dadurch soll ein einfacher Zugang zu Informationen über zugelassene Medizinprodukte sichergestellt werden. Zur Registrierung der Produkte ist eine Single Registration Number (SRN) bei einer benannten Stelle (Competence Authority, CA) zu beantragen. Weiterhin stehen die Hersteller in der Pflicht, die Daten ihrer Produkte in digitaler Form bereitzustellen bzw. diese zu Verwalten. Später müssen diese auf die EUDAMED Datenbank übertragen werden.
<em>Strengere klinische Überwachungen</em>
Mit den Befugnissen der neuen Verordnung haben die Benannten Stellen weitere Kompetenzen in der klinischen Überwachung nach dem Inverkehrbringen. Neu dürfen beispielsweise unangekündigte Audits, Stichproben- und Produktprüfungen durchgeführt werden, was dazu beitragen soll, das Risiko, welches von unsicheren Medizinprodukten ausgeht, zu verringern. Bei definierten Produktgruppen müssen jährlich Berichte über die Sicherheit und Leistung durch die Hersteller eingereicht werden.
<em>Neueinstufung von Produkten</em>
Laut der neuen Verordnung müssen Hersteller die neu definierten Klassifizierungsregeln prüfen und ihre technische Dokumentation dementsprechend anpassen. Die Anpassung an den Klassifizierungsregeln wurden hauptsächlich im Bereich Risiko, Kontaktdauer und Invasivität vorgenommen. Zu berücksichtigen ist zusätzlich, dass für Medizinprodukte der Klasse III und implantierbare Geräte noch strengere klinische Anforderungen gelten. Hier ist eine regelmässige Überwachung zwingend.
Zu der Überprüfung der Einstufung und der Anpassung der technischen Dokumentation gehört auch die Aufteilung in Basic UDI-DI und UDI-DI, welche von der MDR gefordert wird. Die BASIC UDI-DI dient dazu alle gemeinsamen Eigenschaften einer Produktgruppe abzubilden. Die UDI-DI enthält dagegen die produktspezifischen Informationen. Zu einem BASIC UDI-DI kann es mehrere UDI-DIs geben. Umgekehrt ist eine UDI-DI genau einem BASIC UDI-DI zugeordnet.
<em>Strengere klinische Nachweise im Bereich der Klasse III Produkten und implantierbaren Geräten</em>
Wie bereits angesprochen, müssen Hersteller, welche nicht über genügend klinische Nachweise verfügen, um die von der MDR geforderte Sicherheit und Leistung ihres Produkts oder Geräts zu belegen, umfassende klinische Prüfungen durchführen. Zudem besteht die Pflicht unter Mithilfe von laufenden Bewertungen, klinische Daten von Potenziellen Sicherheitsrisiken zu sammeln und aufzubewahren.
<em>Erneute klinische Bewertung von Medizinprodukten der Klassen IIa und IIb</em>
Medizinproduktehersteller müssen ihre klinischen Bewertungen systematisch nochmals erstellen. Dabei müssen sie die neuen Formulierungen in der Verordnung zur Gleichwertigkeit von Produkten berücksichtigt werden. Ebenso müssen die Umstände, unter denen eine klinische Prüfung berechtigterweise entfallen könnte, überprüft werden.
<em>Kein Bestandsschutz</em>
Die Gesetzgebung verlangt eine erneute Zertifizierung aller momentan genehmigten Medizinprodukte nach den neuen Anforderungen. Ob und welche Ausnahmen es geben wird, ist momentan noch in Verhandlung.
<strong>Auswirkungen</strong>
Unter Berücksichtigung der oben genannten Punkte, sind Medizinproduktehersteller gut beraten, wenn sie sich frühzeitig über den Gesetzgebungsprozess der MDR informieren. Man sollte sich regelmässig über die zusätzlichen Änderungen, von denen man betroffen sein könnte, ins Bild setzen und die Umsetzung dieser schnellstmöglich initiieren.
Durch die grosse Menge an Medizinprodukten, welche eine Prüfung sowie eine Freigabe durch eine Benannte Stelle benötigen, ist hier mit Verzögerungen zu rechnen. Verlieren Sie keine Zeit und setzen sie sich zeitnah mit ihrer Benannten Stelle in Verbindung. Somit kann bereits früh eine Lösung konzipiert werden, um eventuelle Probleme bezüglich der Konformität mit der neuen Gesetzgebung zu klären. Eine gute Vorbereitung, sorgfältige Planung und das frühzeitige Veranlassen von Massnahmen sind Kernelemente für eine reibungslose Anpassung an die neuen Anforderungen der Verordnung.</p>
Die Europe IT Consulting GmbH mit Sitz in Basel ist Ihr kompetenter und zuverlässiger Partner bei IT-Lösungen rund um Unique Device Identification (UDI), basierend auf der Branchenlösung SAP ERP. Sie als unser Kunde profitieren von unserer jahrelangen Erfahrung in UDI Projekten.
Mit unseren Add-On Produkten für die Verwaltung und Datenübertagung von UDI Daten wird die Komplexität solcher Projekte auf ein Minimum reduziert. Kompetente Berater unterstützen Sie bei der Durchführung von Projektarbeiten und der zeitgerechten Umsetzung.
Zu unseren Kernkompetenzen zählen die Bereiche:
– Einführung und Beratung von SAP NetWeaver Technologien
– Beratung von Medizintechnik Unternehmen im Bereich UDI und EUDAMED
– Datenverwaltung und Datenübertragung zur FDA’s GUDID von UDI Daten
– Unterstützung bei der UDI Implementierung mit Hilfe unseres UDI SAP Moduls
– ABAP und Webdynpro Entwicklungen
– Fiori / UI5 Entwicklungen
– Formular Entwicklung für SAP (Adobe Forms, Smart Forms, SapScript)
– Inbetriebnahme und Konfiguration von SAP Adobe Document Services (ADS)
– Integration von Scanner- und Druckerlösungen in SAP
Weitere Informationen finden Sie unter www.europe-it-consulting.ch
Europe IT Consulting GmbH
Steinentorstrasse 35
CH4051 Basel
Telefon: +41 (61) 508 73 34
https://www.europe-it-consulting.ch
Geschäftsführer
Telefon: +41 (61) 508 73 34
E-Mail: id@europe-it-consulting.ch
Business Consultant
Telefon: +41 (61) 508 73 34
E-Mail: p.meyer@europe-it-consulting.ch
![]()